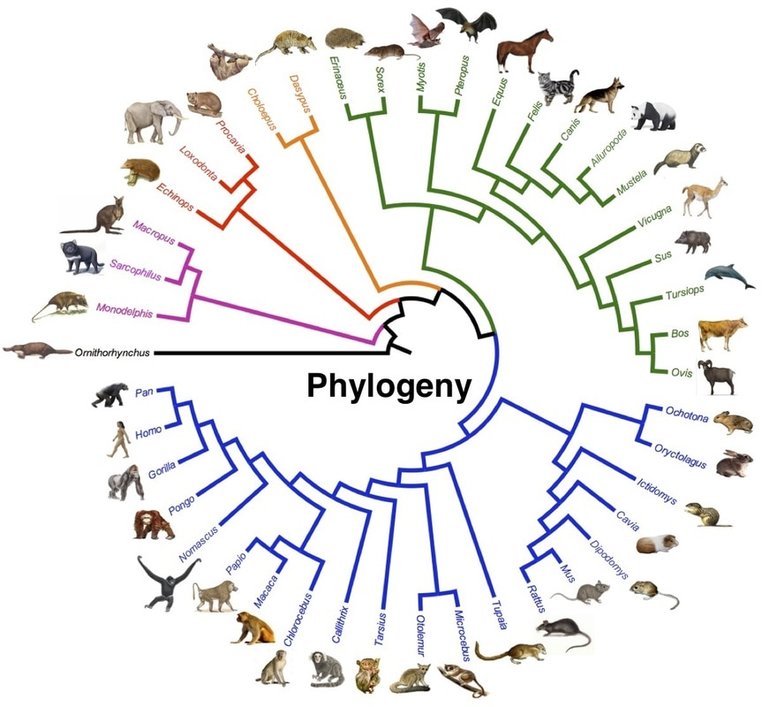
What is Phylogenetics?Phylogenetics is the study of evolutionary relationships between species, populations or individuals [1]. Phylogenetic trees are constructed using molecular data like DNA or protein sequences, enabling us to establish evolutionary events which may have happened in the past [1]. It is extremely useful in a variety of fields, like classification, conservation, identifying the origin of pathogens and many more. Charles Darwin is believed to be the first person to use a tree to represent the branches of evolution (figure 1).
|

Figure 1. Darwin's first phylogenetic tree drawn in On the Orgins of Species (1859)
|
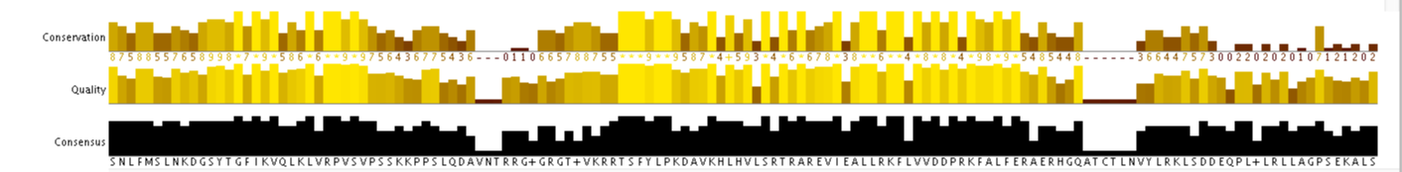
Clustal Omega
Below shows the output from a sequence alignment using protein sequences of RASSF1A homologs.

What methods are used to produce trees?
Distance Matrix: The distance matrix method uses the length of the branches on a tree to indicate the evolutionary distance between species. [2]
Maximum Parsimony: This method produces the simplest tree possible, so there are as few branches as possible. [2]
Maximum likelihood: Likelihood uses biological parameters to determine the probability of there being a change in the sequence. [2]
Bayesian Method: Bayesian is a more computationally demanding method which uses statistics to calculate the most likely tree based on pre-set parameters. [2]
Below shows phylogenetic trees produced from the sequence alignment of RASSF1A homologs. BLOSUM62 is a sequence alignment method which is used to score alignments between evolutionarily divergent protein sequences. Percentage Identity is referring to the percentage identity of similarity between sequences of different homologs and this is used to align the different sequences. Neighbour Joining is a tree building method which joins neighbours from the bottom up based on shared nodes. Some trees are produced using the average (genetic) distance between sequences.
Maximum Parsimony: This method produces the simplest tree possible, so there are as few branches as possible. [2]
Maximum likelihood: Likelihood uses biological parameters to determine the probability of there being a change in the sequence. [2]
Bayesian Method: Bayesian is a more computationally demanding method which uses statistics to calculate the most likely tree based on pre-set parameters. [2]
Below shows phylogenetic trees produced from the sequence alignment of RASSF1A homologs. BLOSUM62 is a sequence alignment method which is used to score alignments between evolutionarily divergent protein sequences. Percentage Identity is referring to the percentage identity of similarity between sequences of different homologs and this is used to align the different sequences. Neighbour Joining is a tree building method which joins neighbours from the bottom up based on shared nodes. Some trees are produced using the average (genetic) distance between sequences.
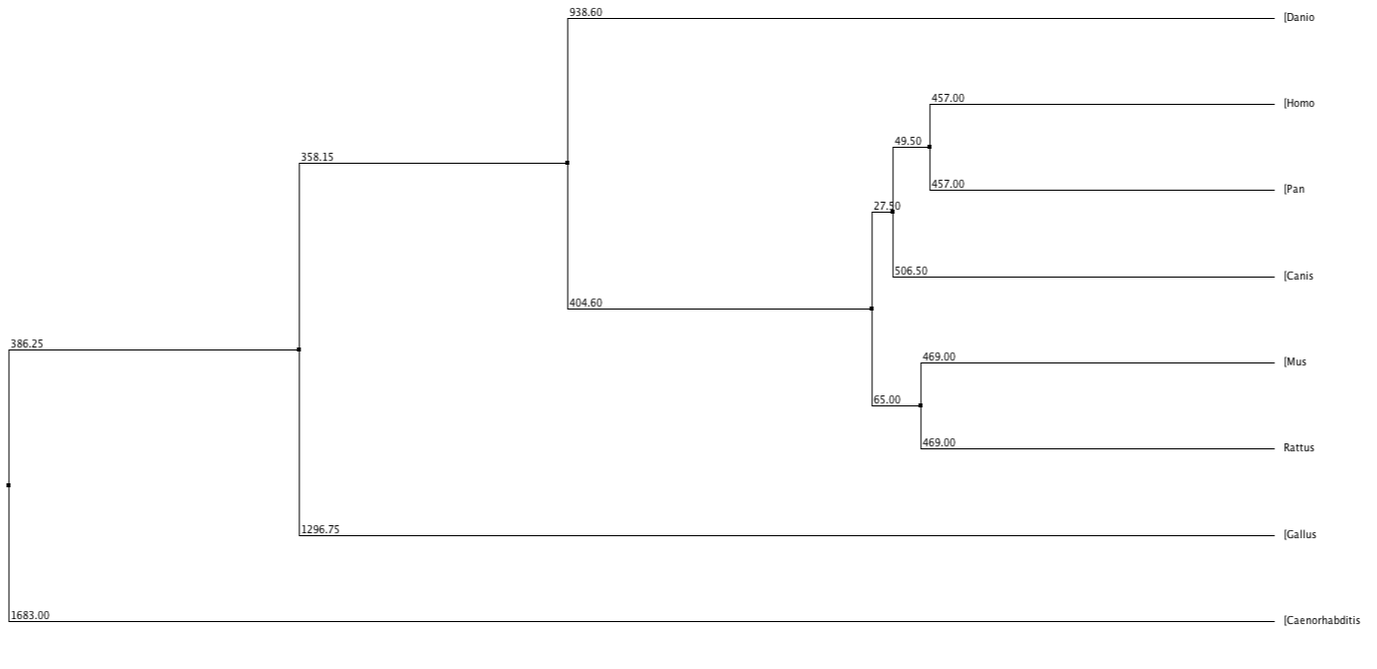
Figure 2. Average Distance tree using BLOSUM62
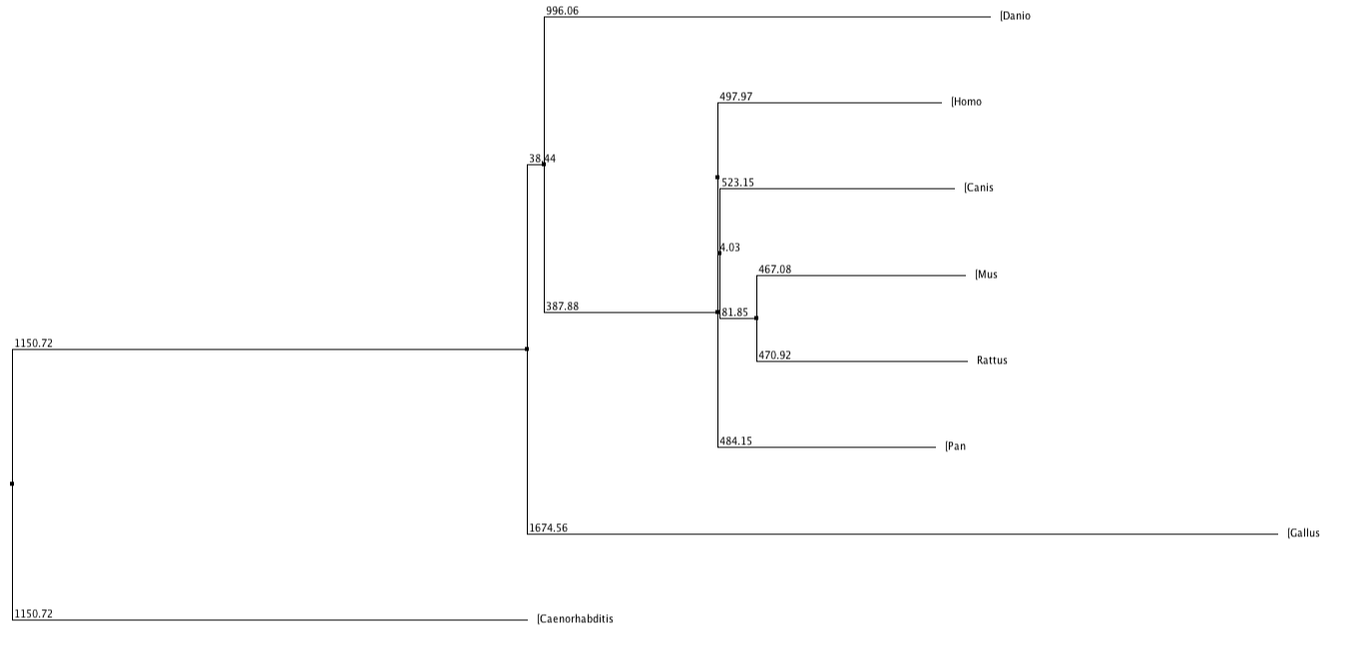
Figure 3. Neighbour Joining tree using BLOSUM62
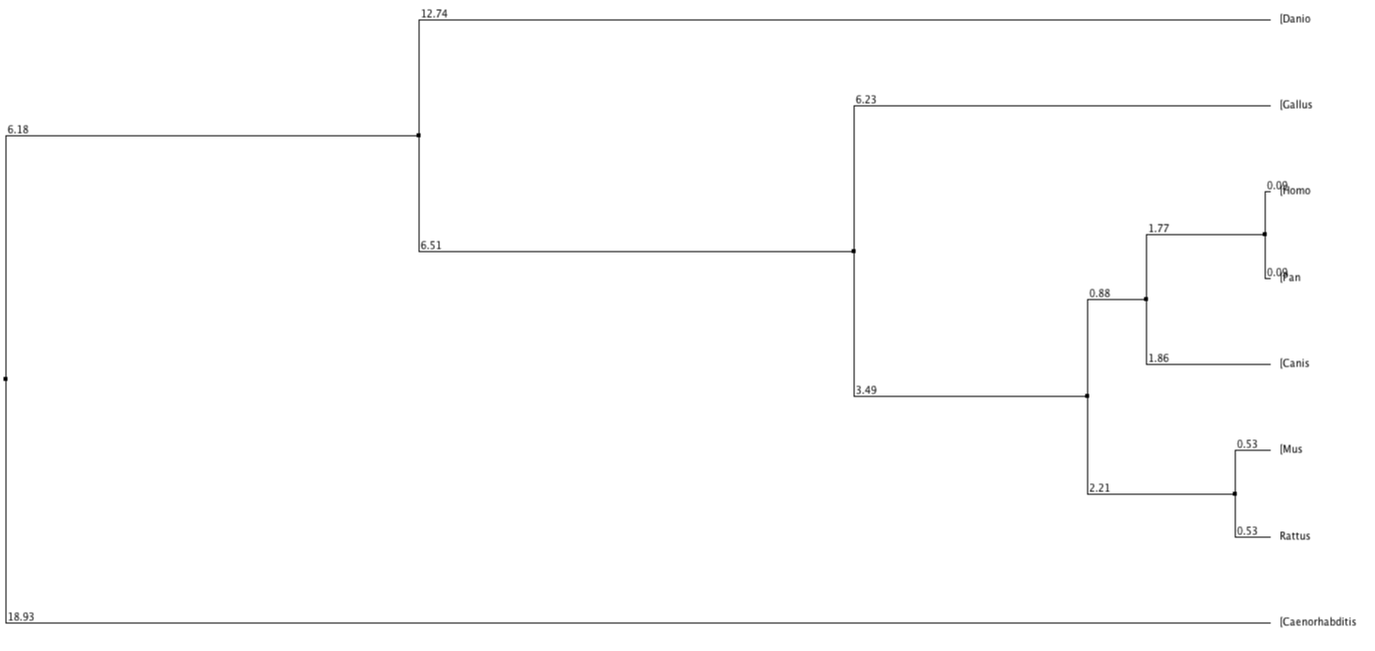
Figure 4. Average Distance tree using Percentage Identity
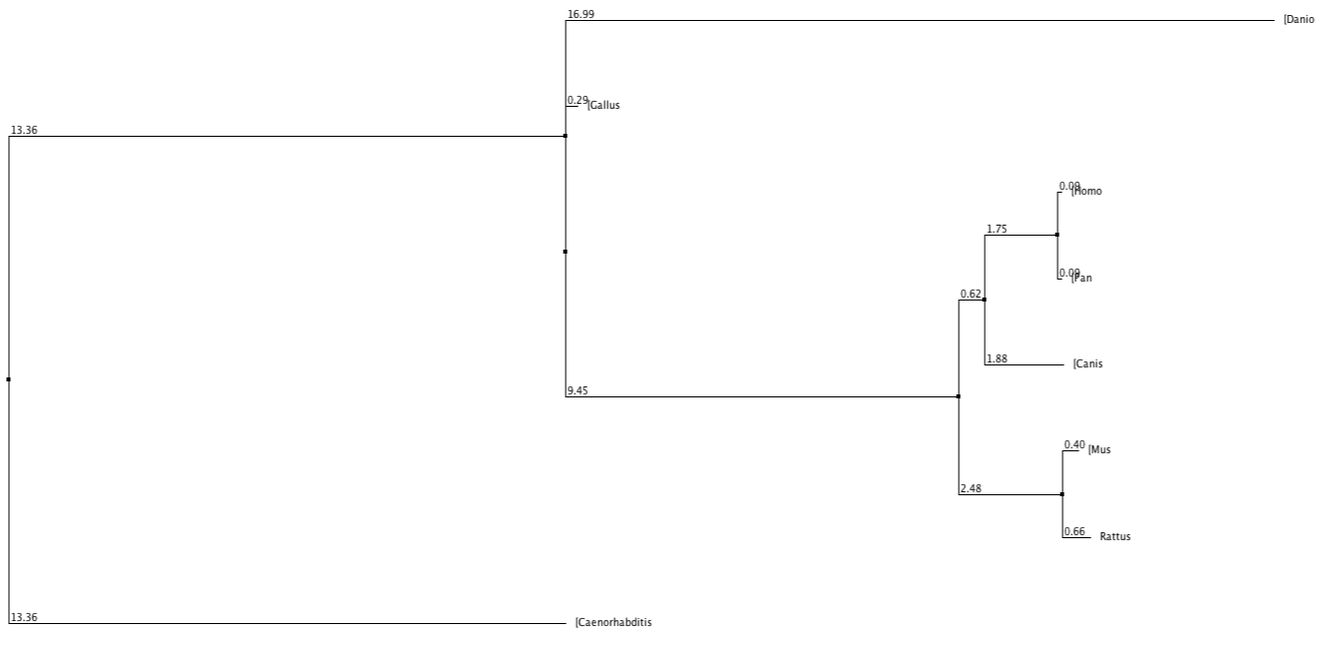
Figure 5. Neighbour Joining tree using Percentage Identity
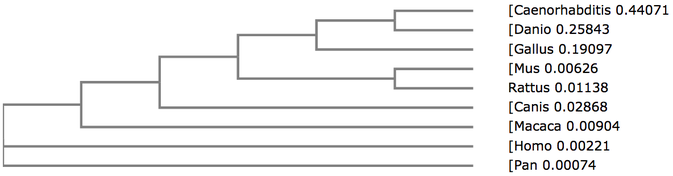
Figure 6. Phylogenetic tree from Clustal Omega
Conclusions
In every tree (except fig. 3 and 6) the chimpanzee can be seen to have exactly the same value, used to measure distance from the last common ancestor, as the human. This shows that they are extremely similar and since the two species have evolved the protein has not changed, much if at all. The nematode forms an out group in every type of tree, suggesting this protein is extremely diverged from any common ancestor.
References
[1] https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[2] Douday, C.J., Delsuc, F., Boucher, Y., Doolittle, F. & Douzery, E. J. P. Comparison of Bayesian and Maximum likelihood bootstrap measures of phylogenetic reliability. 2003. Molecular Biology Evolution. 20 (2): 248-254
[2] Douday, C.J., Delsuc, F., Boucher, Y., Doolittle, F. & Douzery, E. J. P. Comparison of Bayesian and Maximum likelihood bootstrap measures of phylogenetic reliability. 2003. Molecular Biology Evolution. 20 (2): 248-254
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
